Heterocyclen

Heterocyclen (aus altgriechisch ἕτερος héteros „anders, fremd“ und κύκλος kýklos „Kreis“, latinisiert cyclus; Schreibung auch: Heterozyklen und heterocyclische Verbindungen, Singular: der Heterozyklus oder Heterocyclus) sind cyclische chemische Verbindungen mit ringbildenden Atomen aus mindestens zwei verschiedenen chemischen Elementen.[1] Der Begriff wird vorwiegend in der organischen Chemie verwendet und bezeichnet eine ringförmige organische Verbindung, deren Ringgerüst neben Kohlenstoffatomen mindestens ein Atom eines anderen chemischen Elements enthält. Diese Fremdatome werden als Heteroatome bezeichnet. Ein Ringgerüst kann ein oder mehrere gleiche oder verschiedene Heteroatome enthalten. Am häufigsten sind Stickstoff, Sauerstoff und Schwefel vertreten. Aromatische Heterocyclen werden auch kurz als Heteroaromaten bezeichnet.
In der organischen Chemie bilden die Heterocyclen eine umfangreiche Verbindungsklasse. Sie können hierbei nach Art und Anzahl der Heteroatome sowie nach Ringgröße und Sättigungsgrad des cyclischen Systems gegliedert werden. Durch Betrachtung des Sättigungsgrades ergibt sich die Einteilung in gesättigte Heterocycloalkane (vergleiche Cycloalkane), partiell ungesättigte Heterocycloalkene (vergleiche Cycloalkene) und die Heteroaromaten. Besonders in Makrocyclen sind selten auch Cycloalkine (vergleiche Alkine) zu finden.
Geschichte
[Bearbeiten | Quelltext bearbeiten]
Die ersten als Reinsubstanzen erhaltenen heterocyclischen Verbindungen waren Naturstoffe, die im 19. Jahrhundert aus pflanzlichem Material isoliert werden konnten. Auf Grund fehlender analytischer Methoden zu dieser Zeit benötigten Forschungsfortschritte oftmals Jahrzehnte. So wurde erstmals 1806 Morphin von Sertürner in kristalliner Form erhalten, bis zur Bestimmung der korrekten Summenformel der Verbindung vergingen jedoch 42 Jahre und erst 77 Jahre nach seiner Entdeckung konnte die Struktur der Verbindung endgültig geklärt werden.[2] Eine Reihe stickstoffhaltiger heterocyclischer Basen konnte von Thomas Anderson aus tierischem Material gewonnen werden.[3][4][5][6] Zu den isolierten Substanzen gehörten beispielsweise Pyridin und die Picoline, deren Strukturen Jahrzehnte später aufgeklärt werden konnten. Körner postulierte die Hypothese, dass eine Analogie zwischen Benzol und Naphthalin sowie Pyridin und Chinolin bestehe, in den Strukturen der ersteren müsse lediglich eine CH-Einheit durch ein Stickstoffatom ersetzt werden.[7]
Die erste Synthese einer heterocyclischen Verbindung gelang William Ramsay im Jahre 1877. Dieser beobachtete die Bildung von Pyridin bei Durchleitung von Acetylen- und Blausäuregas durch ein rotglühendes Rohr.[8] Im selben Jahr konnten Baeyer und Caro Indol durch die Reaktion von N-Ethylanilin im rotglühenden Rohr erhalten.[9] Bereits zwei Jahre später gelang Königs die erste synthetische Darstellung von Chinolin durch Umsetzung von N-Allylanilin an rotglühendem Bleioxid.[10]
Im Zuge des fortschreitenden Verständnis vom Aufbau heterocyclischer Verbindungen wurden Regeln zu deren Nomenklatur unabhängig voneinander von Arthur Hantzsch[11] und Oskar Widman[12] in den Jahren 1887 und 1888 aufgestellt. Diese wurden im Laufe der Jahre erweitert, bilden jedoch immer noch das Grundgerüst der heute gängigen Hantzsch-Widman-Nomenklatur.
Nomenklatur, Stammsysteme und Verbindungen
[Bearbeiten | Quelltext bearbeiten]Zur Nomenklatur heterocyclischer Systeme wurde von der IUPAC die Verwendung des Hantzsch-Widman-Systems empfohlen.[13] Es sind jedoch noch weitere Nomenklaturen gebräuchlich; umgangssprachlich werden oftmals Trivialnamen verwendet.[2]
Heterocyclen mit Elementen der fünften und sechsten Hauptgruppe
[Bearbeiten | Quelltext bearbeiten]| Einfache Heterocyclen mit einem Heteroatom[13][11][12] | ||||||
|---|---|---|---|---|---|---|
| Gesättigte Heterocyclen | Ungesättigte Heterocyclen | |||||
| Heteroatom | Stickstoff | Sauerstoff | Schwefel | Stickstoff | Sauerstoff | Schwefel |
| 3-Ringe | ||||||
| Nomenklaturname | Aziridin | Oxiran | Thiiran | Azirin | Oxiren | Thiiren |
| Trivialname | Ethylenimin | Ethylenoxid | Ethylensulfid | – | Acetylenoxid | Acetylensulfid |
| Struktur |  |
 |
 |
 |
 |

|
| 4-Ringe | ||||||
| Nomenklaturname | Azetidin | Oxetan | Thietan | Azet | Oxetiumion | Thietiumion |
| Trivialname | 1,3-Propylenimin | Trimethylenoxid | Trimethylensulfid | Azacyclobutadien | – | – |
| Struktur |  |
 |
 |
 |
 |

|
| 5-Ringe | ||||||
| Nomenklaturname | Azolidin | Oxolan | Thiolan | Azol | Oxol | Thiol |
| Trivialname | Pyrrolidin | Tetrahydrofuran | Tetrahydrothiophen | Pyrrol | Furan | Thiophen |
| Struktur |  |
 |
 |
 |
 |

|
| 6-Ringe | ||||||
| Nomenklaturname | Azinan | Oxan | Thian | Azin | Oxiniumion | Thiiniumion |
| Trivialname | Piperidin | Tetrahydropyran | Tetrahydrothiopyran | Pyridin | Pyryliumion | Thiopyryliumion |
| Struktur |  |
 |
 |
 |
 |

|
| 7-Ringe | ||||||
| Nomenklaturname | Azepan | Oxepan | Thiepan | Azepin | Oxepin | Thiepin |
| Trivialname | Hexamethylenimin | Hexamethylenoxid | Hexamethylensulfid | Azatropiliden | Oxacycloheptatrien | Thiotropiliden |
| Struktur |  |
 |
 |
 |
 |

|
Auch eine Reihe weiterer Heterocyclen ist von großer Bedeutung und weitverbreitet anzutreffen. Hierzu gehören sowohl Verbindungen, welche mehrere Heteroatome tragen, als auch solche die aus mehrkernigen Ringsystemen bestehen. Die prominentesten Stammsysteme bilden fünfgliedrige stickstoffhaltige ungesättigte Heterocyclen wie Pyrazol, Imidazol und Indol sowie die sechsgliedrigen Ringe Chinolin, Isochinolin, Purin, Pyrimidin und das gesättigte Piperidin. Des Weiteren sind auch Oxazole, Thiazole und Thiazine häufig anzutreffen.












Während der Großteil der heterocyclischen Verbindungen Sauerstoff, Stickstoff oder Schwefel als Heteroatome trägt, sind seltener auch weitere Elemente zu finden. So sind Phosphabenzol und Arsabenzol als Homologe des Pyridins bekannt.[14] Auch Phosphol und Arsol als Pyrrolhomologe konnten synthetisiert werden.[15]



Metallacyclen
[Bearbeiten | Quelltext bearbeiten]Nicht nur Hauptgruppenelemente können als Heteroatome fungieren, auch Nebengruppenelemente können in cyclische Systeme eingebaut sein. Beispiele sind das Cyclopentenderivat in Hoveyda-Katalysatoren, das neben einem Sauerstoff- auch ein Rutheniumatom enthält. In der Pauson-Khand-Reaktion sowie in der Nicholas-Reaktion werden Dicobaltderivate des Cyclobutans gebildet.[16]
Eine Besonderheit sind die Metallabenzole und Metallabenzine. Hierbei handelt es sich um ungesättigte sechsgliedrige Heterocyclen, die Fragmente von Übergangsmetallkomplexen in ihrer Ringstruktur tragen. Die Existenz solcher Systeme wurde 1979 von Thorn und Hoffmann vorhergesagt.[17] Als erste Verbindung dieser Klasse konnte drei Jahre später ein Osmabenzol dargestellt werden.[17] Es konnte gezeigt werden, dass es sich hierbei um ein aromatisches System handelt, das Substitutionsreaktionen ähnlich Benzol eingeht.[18] Analog zu Benzin konnten auch Metallabenzine dargestellt werden, die eine Dreifachbindung im Sechsring tragen. Im Gegensatz zu Benzin gehen diese jedoch keine Additionsreaktionen an der Dreifachbindung ein.[19]
-
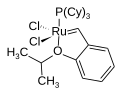 Struktur des Hoveyda-Katalysators I; Cy = Cyclohexyl
Struktur des Hoveyda-Katalysators I; Cy = Cyclohexyl -
 Struktur des ersten Metallabenzols
Struktur des ersten Metallabenzols -
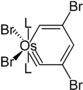 Struktur eines Osmabenzins
Struktur eines Osmabenzins



- →Für weitere heterocyclische Verbindungen siehe auch Kategorie:Heterocyclische Verbindung.
Vorkommen
[Bearbeiten | Quelltext bearbeiten]Heterocyclische Verbindungen sind in der Natur eine weitverbreitete Verbindungsklasse. Sie besitzen wichtige Funktionen in biologischen Prozessen, die sie meist auf Grund ihrer Fähigkeit zur Komplexbildung, seltener auch auf Grund ihrer Brønsted-Basizität, übernehmen. Im Falle der Alkaloide, zu deren prominentesten Vertretern Atropin, Kokain, Koffein und Nicotin sowie die Opiate gehören, treten sie als natürliche Giftstoffe auf, die auch als Rauschmittel missbraucht werden können. Einige Mitglieder dieser Gruppe finden in der Medizin Anwendung.[2][20] Auch drei der natürlichen Aminosäuren, namentlich Prolin (Pyrrolidinderivat), Histidin (Imidazolderivat) und Tryptophan (Indolderivat), besitzen heterocyclische Ringe.
DNA
[Bearbeiten | Quelltext bearbeiten]
In der DNA wird die Aufgabe der Nucleinbasen von heterocyclischen Verbindungen übernommen. Die Nucleinbasen der DNA sowie auch der RNA besitzen einen Purin- (Adenin, Guanin, Xanthin und Hypoxanthin) beziehungsweise Pyrimidingrundkörper (Cytosin, Thymin und Uracil). Ihre basischen Eigenschaften erlauben die Ausbildung von Wasserstoffbrückenbindungen zwischen den komplementären Basenpaaren und bilden so die Bindungen zwischen den beiden Einzelsträngen der DNA.[21] Eine medizinische Anwendung ergibt sich aus der Fähigkeit der Nucleinbasen zur Komplexbildung. So beruht beispielsweise die Wirkung des Zytostatikums Cisplatin, das zur Krebstherapie eingesetzt wird, auf der Komplexierung des eingesetzten Platinkomplexes an das N7-Atom von Guanin und Adenin. Die hieraus resultierenden Vernetzungen zwischen den einzelnen Strängen bewirken eine Behinderung des Zellstoffwechsels und führen meist zum Zelltod, wodurch die Reproduktion von Krebszellen unterbunden wird.[22][23] Auch die Koenzyme NAD, NADP und ATP besitzen diese Grundkörper.
Cytochrome
[Bearbeiten | Quelltext bearbeiten]
Unter Cytochromen wird die Gruppe der farbigen Proteine zusammengefasst, welche Häme als prosthetische Gruppen besitzen. Diese treten in einigen biologisch bedeutsamen Prozessen auf. Hierzu gehören der Sauerstofftransport, der im Blut durch den roten Blutfarbstoff Hämoglobin und im Muskelgewebe durch das Myoglobin bewerkstelligt wird. Bei Photosynthese und Zellatmung treten sie ebenso in Erscheinung wie in vielen Enzymen, wie beispielsweise in der Cytochrom-P450-Superfamilie. Die Hämgruppe ist aus Derivaten des Porphyrins aufgebaut, welches verschiedene Metallionen komplexiert. Porphyrine sind makrocyclische Verbindungen, die sich formal aus vier Pyrroleinheiten zusammensetzen.[24][25] Der Makrocyclus tritt hierbei als vierzähniger Chelat-Ligand auf und vermag auf Grund des makrocyclischen Effekts auch mit schlecht zu komplexierenden Ionen wie Magnesium thermodynamisch stabile, jedoch kinetisch labile Komplexe zu bilden. Durch dieses variable System ist gewährleistet, dass je nach Anforderung eine Vielzahl von Ionen Verwendung finden kann. Im Falle des Hämoglobins und des Myoglobins handelt es sich hierbei um Eisenionen, während im nicht zu den Cytochromen gehörenden Chlorophyll ein Magnesiumkation gebunden ist.[26]
Cobalamine
[Bearbeiten | Quelltext bearbeiten]
Die Gruppe der Cobalamine, wichtige Coenzyme mit ihrem prominentesten Vertreter dem Vitamin B12, sind Cobaltkomplexe, deren Metallion durch einen makrocyclischen Corrinring komplexiert wird. Ähnlich dem Porphyrinring ist auch der Corrinring formal aus vier Pyrroleinheiten zusammengesetzt, ist jedoch um ein Brückenglied kürzer. Dies führt zu einem engeren Ringöffnung, was die Komplexierung kleinerer Metallionen erlaubt. Eine axiale Koordinationsstelle wird durch einen Histidinliganden besetzt. Der Heteromakrocyclus übernimmt hier also abermals die Rolle eines Komplexbildners, der sowohl thermodynamisch stabile als auch kinetisch inerte Komplexe bildet.[27] Er bleibt hierbei jedoch so flexibel, dass er Ionen eines ganzen Größenbereichs aufzunehmen vermag, so in diesem Fall sowohl mono-, als auch di- und trivalentes Cobalt.[28] Die Flexibilität wird durch die Verdrillung des planaren Liganden in der Ebene beziehungsweise durch dessen Wölben (doming) erreicht.[27]
Natürliche Farb- und Geruchsstoffe
[Bearbeiten | Quelltext bearbeiten]

Indigo ist ein pflanzlicher Farbstoff, der schon vor über 4000 Jahren zum Färben von Stoffen verwendet wurde. Er wird aus Pflanzen wie der Indigofera arrecta, Indigofera tinctoria oder der Indigofera suffruticosa gewonnen. Diese enthalten jedoch kein Indigo, sondern Indican, ein Glucosid des Indoxyls, aus dem auf chemischem Wege Indigo gewonnen wird. Dies geschieht durch Hydrolyse des Glucosids, wodurch zunächst das gelbe Indoxyl gebildet wird, welches anschließend oxidativ zu Indigo umgesetzt wird.[29] Sowohl Indigo als auch das Indican besitzen ein Indol-Grundgerüst. Indican selbst ist farblos, Indigo selbst besitzt eine charakteristische blaue Farbe.

Bei Skatol handelt es sich um ein weiteres Indolderivat, nämlich 3-Methylindol. Skatol ist einer der Hauptgeruchsstoffe des menschlichen und tierischen Kots. Auch Zibet, das zur Parfümherstellung verwendet wird, enthält geringe Mengen Skatol.[30] Der Stoff besitzt eine sehr niedrige Geruchsschwelle und kann somit noch in sehr hoher Verdünnung wahrgenommen werden. So sind 0,23 µg pro kg Stärke für den Menschen noch wahrnehmbar.[31] Die Biosynthese verläuft über eine zweistufige Oxidation von Tryptophan mit anschließender Decarboxylierung.[31] Da tierisches Gewebe eine höhere Tryptophankonzentration besitzt, riecht der Kot fleischfressender Tiere meist strenger als der pflanzenfressender Arten.
Eigenschaften
[Bearbeiten | Quelltext bearbeiten]Über die chemischen und physikalischen Eigenschaften von Heterocyclen können keine allgemeingültigen Aussagen getroffen werden, sie können jedoch gruppenweise nach Art des Heteroatoms und dem Sättigungsgrad eingeteilt und behandelt werden. Es bietet sich an, hierbei zwischen aromatischen und ungesättigten Heterocyclen zu unterscheiden.
Aromatizität
[Bearbeiten | Quelltext bearbeiten]
Die Aromatizität spiegelt die Eigenschaft aromatischer Verbindungen wider, ein ringförmiges System aus delokalisierten π-Elektronen auszubilden. Ebenso wie das Benzol als klassisches Beispiel eines Aromaten, besitzen die meisten ungesättigten Heterocyclen, die die Hückel-Kriterien erfüllen, aromatische Eigenschaften.[2][32] Das Kation des Azirins ist der einfachste aromatische Heterocyclus. Er besitzt lediglich ein π-Elektronenpaar und erhält durch die Delokalisation der positiven Ladung seinen aromatischen Charakter. Dahingegen ist das entsprechende Anion ein klassischer Antiaromat.

Bei Hückel-Heteroaromaten ist die Aromatizität stark von der Elektronegativität der Heteroatome abhängig. Besitzen diese eine hohe Elektronegativität, so kommt es zur Lokalisation eines höheren Teils der Elektronendichte an diesen Kernen, wodurch sich die Aromatizität des Systems verringert. Dies kann beispielsweise an der Reihe der fünfgliedrigen Heterocyclen beobachtet werden. Während das schwefelhaltige Thiophen sich den aromatischen Eigenschaften von Benzol ähnelt, nehmen diese mit ansteigender Elektronegativität des Heteroatoms über Pyrrol zu Furan stark ab.[2][32][33] Die Aromatizität des Furans ist aufgrund einer starken Lokalisierung der Elektronen so schwach, dass es selbst Diels-Alder-Reaktionen eingeht, ein für Aromaten untypisches Verhalten, da die Reaktion mit dem Verlust der Mesomeriestabilisierung einhergeht.[2]
Basizität
[Bearbeiten | Quelltext bearbeiten]Häufig anzutreffenden Heteroatome besitzen in der Regel mindestens ein freies Elektronenpaar. Hierdurch erhalten sie sowohl einen Brønsted- als auch einen Lewis-basischen Charakter.[2] So besitzt Pyridin bei 20 °C einen pKb-Wert von 8,94.[34] Sind mehrere elektronenziehende Heteroatome vorhanden, so verringert sich die Basizität, da diese um Elektronendichte konkurrieren und dadurch nur einen geringeren Betrag auf sich selbst vereinen können. Dementsprechend weisen sechsgliedrige Heteroaromaten, die zwei Stickstoffe im Ring tragen, höhere pKb-Werte auf als Pyridin (Pyridazin 11,71, Pyrimidin 12,87, Pyrazin 13,6 bei 20 °C).[34]
Auf Grund ihrer Lewis-Basizität können heterocyclische Verbindungen auch als Liganden in Komplexen fungieren. Gesättigte Heterocyclen sind in der Regel klassische σ-Donor-Liganden, während Heteroaromaten auch π-Akzeptor-Liganden darstellen. Durch die Fähigkeit der Ausbildung von Rückbindungen vom Metallzentrum in das LUMO des Liganden, bewirken sie meist eine hohe Ligandenfeldaufspaltung und bilden stabile Komplexe.[35][18]
Synthese
[Bearbeiten | Quelltext bearbeiten]Stickstoffhaltige Heterocyclen
[Bearbeiten | Quelltext bearbeiten]Azirine können auf photochemischem Wege aus Vinylaziden in guter Ausbeute gewonnen werden.[36] Die gesättigten Aziridine sind über eine intramolekulare Substitution zugänglich. Hierzu werden Amine, die eine Abgangsgruppe in der β-Position besitzen, beispielsweise 2-Bromethylamin, im basischen Milieu eingesetzt.

Pyrrolderivate sind auf einfachem Wege durch die Paal-Knorr-Synthese erhältlich.[37][32] Hierbei werden 1,4-Dicarbonylverbindungen mit Ammoniak beziehungsweise einem primären Amin zur Reaktion gebracht. Die gesättigten und teilweise ungesättigten Pyrrolidine sind aus diesen meist durch Hydrierung zugänglich.
Für die häufig anzutreffende Gruppe der Pyridine existiert die Hantzschsche Pyridinsynthese als einfacher synthetischer Zugangsweg.[38] Hierbei handelt es sich um eine Abfolge von Kondensationsreaktionen und Isomerisierungen, durch welche aus Ketonen, Aldehyden und Ammoniak zunächst eine Dihydropyridin-Spezies gebildet wird, welche dann mittels Salpetersäure zum Pyridinderivat oxidiert werden kann.[2]
Sauerstoffhaltige Heterocyclen
[Bearbeiten | Quelltext bearbeiten]Zur Synthese von Epoxiden wird meist die Prileschajew-Reaktion angewandt.[39] Diese geht aus von Alkenen, die mittels Peroxycarbonsäuren oxidiert werden. Hierzu werden mit elektronenziehenden Gruppen substituierte Persäuren verwendet, meist meta-Chlorperbenzoesäure. Die enantioselektive Synthese von Epoxiden kann mit Hilfe der Sharpless-Epoxidierung durchgeführt werden, für deren Entwicklung unter anderem 2001 der Nobelpreis für Chemie an Barry Sharpless verliehen wurde.[40]
Oxetane können durch die Paternò-Büchi-Reaktion synthetisch erhalten werden.[41] Zu dieser [2+2]-Cycloaddition wird ein Alken mit ein einer Carbonylverbindung photochemisch zur Reaktion gebracht.[42]
Das jährlich im Multi-Tonnen-Maßstab hergestellte Furan kann großtechnisch aus der Reaktion zwischen Acetylen und Formaldehyd und anschließender Umsetzung des so erhaltenen Diols mit Ammoniak gewonnen werden.[43] Das gesättigte Tetrahydrofuran (THF) sowie teilweise ungesättigte Derivate können aus den gebildeten Furanderivaten durch Hydrierung dargestellt werden.
Schwefelhaltige Heterocyclen
[Bearbeiten | Quelltext bearbeiten]Thiirene als einfachste ungesättigte schwefelhaltige dreigliedrige Heterocyclen sind instabil und aus diesem Grund nur sehr selten anzutreffen. Sie können beispielsweise durch eine photochemische Reaktion aus Thioacetaldehyd erhalten werden.[44] Für die gesättigten Thiirane hingegen ist eine Vielzahl von Syntheserouten in der Literatur beschrieben. Ein einfacher Zugang ist durch eine Substitutionsreaktion von Sulfiden an α,β-Dibromalkanen gegeben.[45]
3-Thiazoline lassen sich in einer Mehrkomponentenreaktion – der Asinger-Reaktion[46] oder davon abgeleiteten Reaktionen[47] – synthetisieren.
Thiophene sowie deren gesättigte Derivate sind die häufigsten schwefelhaltigen Heterocyclen. Wie auch dessen Stickstoff- und Sauerstoffanaloga kann Thiophen durch eine Paal-Knorr-Synthese aus einer 1,4-Dicarbonylverbindung gewonnen werden. Das Schwefelatom wird in diesem Fall durch die Verwendung von Schwefelwasserstoff oder Phosphorpentasulfid eingeführt.[32] Substituierte Thiophene können mittels der Gewald-Reaktion synthetisiert werden.[48]
Reaktionen
[Bearbeiten | Quelltext bearbeiten]Aromatische Substitution
[Bearbeiten | Quelltext bearbeiten]
Da bei elektrophilen aromatischen Substitutionen zunächst ein π-Komplex zwischen Elektrophil und Aromat ausgebildet wird, ist der Erfolg der Reaktion abhängig von der π-Elektronendichte des Aromaten. In Heteroaromaten, in denen das freie Elektronenpaar des Heteroatoms nicht am π-Systems beteiligt ist, ist die π-Elektronendichte im Vergleich zum reinen Kohlenstoffaromaten in der Regel niedriger. Daher ist die Bildung des π-Komplexes weniger bevorzugt und eine elektrophile Substitution erschwert (elektronenarme Heteroaromaten, Beispiel: Pyridin).[32] Hingegen ist die Elektrondichte erhöht, wenn das freie Elektronenpaar des Heteroatoms am π-Systems beteiligt ist, und damit die elektrophile Substitution erleichtert (Elektronenüberschuss-Heteroaromat, Beispiel: Pyrrol).[49]
Bezüglich der Regioselektivität können Heteroatome elektronenarmer Heteroaromaten ähnlich wie Substituenten mit elektronenziehendem Charakter behandelt werden. Dies bedeutet in Bezug auf die sechsgliedrigen Heteroaromaten, dass eine elektrophile Substitution bevorzugt in der β-Position zum Heteroatom ablaufen wird. Die Begründung hierfür ist jedoch weniger in der Elektronendichte an dieser Position zu suchen, sondern liegt zum Großteil darin begründet, dass so im intermediären σ-Komplex keine mesomere Grenzstruktur existiert, die eine positive Ladung am elektronegativen Heteroatom besitzt.[50][51] Analog lässt sich begründen, dass elektronenreiche Heteroaromaten bevorzugt in α-Position zum Heteroatom substituiert werden.
Schwache Elektrophile reagieren mit elektronenarmen Heteroaromaten meist schlecht. Systeme mit stark verminderter Elektronendichte, zum Beispiel solche mit mehreren Heteroatomen oder elektronenziehenden Substituenten, reagieren oft gar nicht im Sinne einer elektrophilen Substitution, sondern nur in einer nucleophilen aromatischen Substitution. Ein Beispiel hierfür ist die Tschitschibabin-Reaktion zur Synthese von α-Aminopyridinen.[52][53]
Ringöffnung
[Bearbeiten | Quelltext bearbeiten]
Durch Reaktion mit geeigneten Nucleophilen können einige meist gesättigte Heterocyclen geöffnet werden. Diese Ringöffnung wird durch die Gegenwart von Lewis-Säuren begünstigt. Ein Beispiel hierfür ist die Öffnung von Thietan durch Brom.[54]

Durch ein Nukleophil können auch Epoxide geöffnet werden. Ist das entstandene Alkoholat nicht ausreichend stabilisiert, so fungiert es als Nucleophil für weitere Ringöffnung. Es folgt eine Polymerisation unter Bildung eines Polyethers.[55]
Die Öffnung von Epoxiden wird bei einer Reihe chemischer Reaktionen ausgenutzt. So wird das Epoxid bei der Dihydroxylierung zur Herstellung von trans-Diolen mit Natron- oder Kalilauge geöffnet,[56] eine intramolekulare Öffnung durch Elektronenverschiebung wird bei der Eschenmoser-Fragmentierung ausgenutzt.[57][58]
Verwendung
[Bearbeiten | Quelltext bearbeiten]
Heterocyclische Verbindungen sind praktisch überall anzutreffen, so dass eine ausführliche Betrachtung mehrere Buchbände füllen würde. Der Großteil pharmazeutischer Wirkstoffe ist aus Heterocyclen aufgebaut.[59] Zu den meistverkauften Präparaten zählen Atorvastatin (Sortis®, Lipitor®), Sildenafil (Viagra®) und Diazepam (Valium®). Das gesamte jährliche Verkaufsvolumen dieser Medikamente beläuft sich auf fast 100 Milliarden Euro.[60]
In der präparativen Chemie
[Bearbeiten | Quelltext bearbeiten]Als Basen
[Bearbeiten | Quelltext bearbeiten]
In der präparativen Chemie dienen vorwiegend aromatische oder teilweise gesättigte stickstoffhaltige Heterocyclen als Basen. Sie werden häufig zur Deprotonierung von Alkoholen oder stark C,H-acider Verbindungen eingesetzt.[32] Neben Pyridin ist dies das Hauptanwendungsgebiet der Basen DBU und DBN. Ihr Vorteil liegt in der Basenstärke, die im Vergleich zu organometallischen Basen wie den Lithiumalkylen oder zu Hydriden schwächer ist. Hierdurch wird eine höhere Selektivität gewährleistet. Des Weiteren sind sie meist nur schwache Nucleophile, sie greifen also Carbonylverbindungen in der Regel nicht an, was die Auswahl der möglichen Substrate erhöht.
Als Lösungsmittel
[Bearbeiten | Quelltext bearbeiten]
Unter den häufig verwendeten Lösungsmitteln sind auch heterocyclische Verbindungen zu finden. Hierzu gehört vor allem Tetrahydrofuran (THF), das als schwach polarer und schwach koordinierender Ether Anwendung findet. Auch Pyridin und sehr selten auch Pyrimidin und Piperidin werden als basische Lösungsmittel eingesetzt. Einen großtechnischen Einsatz des Schwefelcyclus Sulfolan wurde vom Mineralölunternehmen Shell in den 1960er Jahren entwickelt. Dieses wird zur Reinigung von Rohprodukten von aromatischen Verbindungen in einem flüssig-flüssig-extraktiven Verfahren genutzt.[61]
In der Elektrotechnik
[Bearbeiten | Quelltext bearbeiten]
Heterocyclische Verbindungen können stromleitende Eigenschaften besitzen und somit als Ersatz für herkömmliche metallische Leiter in elektronischen Bauteilen eingesetzt werden. Sie können als elektrische Leiter, Halbleiter oder Supraleiter beispielsweise in Batterien, Transistoren und Leuchtdioden verwendet werden.[60] Hierzu eignen sich Verbindungen, die ein ausgedehntes konjugiertes π-Elektronensystem besitzen, wie etwa Polythiophene oder Polypyrrole.[62][63] Interessante Eigenschaften haben auch Einkristalle aus Tetrathiafulvalen (TTF) und Tetracyanochinodimethan (TCNQ) gezeigt.[64][65]
Literatur
[Bearbeiten | Quelltext bearbeiten]Allgemeine Lehrbücher der Organischen Chemie mit Kapiteln zur Heterocyclenchemie:
- Tanja Schirmeister, Carsten Schmuck, Peter R. Wich, Hans Beyer, Wolfgang Walter: Lehrbuch der Organischen Chemie. 25. Auflage. S. Hirzel Verlag, Stuttgart 2015, ISBN 978-3777616735, S. 753–941.
- Stuart Warren, Jonathan Clayden, Nick Greeves: Organische Chemie, 2. Auflage, Springer, 2013, ISBN 978-364234715-3, S. 796–933.
- K. Peter C. Vollhardt, Neil E. Schore, Holger Butenschön, Organische Chemie, 6. Auflage, Wiley-VCH, 2020, ISBN 978-352734584-7, S. 1354–1404.
- Julio Alvarez-Builla, Juan Jose Vaquero, José Barluenga: Modern Heterocyclic Chemistry. 1. Auflage, Wiley-VCH, 2011, ISBN 9783527332014.
Spezielle Lehrbücher zur Chemie von Heterocyclen:
- Theophil Eicher, Siegfried Hauptmann, Andreas Speicher: The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, Second Edition. 3. Auflage, Wiley-VCH, 2012, ISBN 9783527307203.
- John A. Joule, Keith Millis: Heterocyclic Chemistry. 5. Auflage, Wiley-VCH, 2010, ISBN 978-1405133005.
- Alan R. Katritzky, Christopher A. Ramsden, John A. Joule, Viktor V. Zhdankin: Handbook of Heterocyclic Chemistry. 3. Auflage, Elsevier, 2010, ISBN 978-0080958439.
- Hans Neunhoeffer, Thomas L Gilchrist: Heterocyclenchemie. Wiley-VCH, ISBN 978-3527292233.
- David T. Davies: Basistexte Chemie: Aromatische Heterocyclen. 1. Auflage. Wiley-VCH, Weinheim 1995, ISBN 3-527-29289-6.
- Josef Houben, E. Pfankuch: Fortschritte der Heilstoff-Chemie. Zweite Abteilung: Die Ergebnisse der wissenschaftlichen Literatur. 4 Bände. 1930 ff., Band 3: Die heterocyclischen Verbindungen mit Ring-Sauerstoff- und einem oder zwei Ring-Stickstoffatomen. Walter de Gruyter & Co., Berlin 1939.
Weblinks
[Bearbeiten | Quelltext bearbeiten]- Vorlesungsskript Universität Tübingen ( vom 12. April 2015 im Internet Archive) (PDF-Datei; 1,20 MB)
- Vorlesungsskript Universität Regensburg ( vom 29. Juni 2007 im Internet Archive) (PDF-Datei; 1,97 MB)
Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ Eintrag zu heterocyclic compounds. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.H02798 – Version: 2.1.5.
- ↑ a b c d e f g h H. Beyer, W. Walter: Lehrbuch der Organischen Chemie. 23. Auflage. S. Hirzel Verlag, Stuttgart 1998, ISBN 3-7776-0808-4, S. 753–876.
- ↑ Th. Anderson: Ueber Picolin. Eine neue Basis aus dem Steinkohlen-Theeröl. In: Liebigs Ann. 60, 1846, S. 86–103; doi:10.1002/jlac.18460600106.
- ↑ Th. Anderson: Producte der trocknen Destillation thierischer Materien. In: Liebigs Ann. 70, 1849, S. 32–38; doi:10.1002/jlac.18490700105.
- ↑ Th. Anderson: Ueber die Producte der trocknen Destillation thierischer Materien. In: Liebigs Ann. 80, 1851, S. 44–65; doi:10.1002/jlac.18510800104.
- ↑ Th. Anderson: Ueber die Producte der trocknen Destillation thierischer Materien. In: Liebigs Ann. 94, 1855, S. 358–365; doi:10.1002/jlac.18550940312.
- ↑ A. Ladenburg: Lectures on the history of the development of chemistry since the time of Lavoisier. Englische Übersetzung einer Vorlesung. Volltextzugriff (PDF; 5,0 MB).
- ↑ Über W. Ramsays Entdeckung in: Ber. Dtsch. Chem. Ges. 10, 1877, S. 736; doi:10.1002/cber.187701001202.
- ↑ A. Baeyer, H. Caro: Indol aur Äthylanilin. Ber. Dtsch. Chem. Ges. 10, 1877, S. 692–693; doi:10.1002/cber.187701001193.
- ↑ W. Königs: Synthese des Chinolins aus Allylanilin. In: Ber. Dtsch. Chem. Ges. 12, 1879, S. 453; doi:10.1002/cber.187901201128.
- ↑ a b A. Hantzsch, J. H. Weber: Ueber Verbindungen des Thiazols (Pyridins der Thiophenreihe). In: Ber. Dtsch. Chem. Ges. 20, 1887, S. 3118–3132; doi:10.1002/cber.188702002200.
- ↑ a b O. Widman: Zur Nomenclatur der Verbindungen, welche Stickstoffkerne enthalten. In: J. Prakt. Chem. 38, 1888, S. 185–201; doi:10.1002/prac.18880380114.
- ↑ a b W. H. Powell: Revision of the extended Hantzsch-Widman System of Nomenclature for Heteromonocycles. In: Pure Appl. Chem. 55, 1983, S. 409–416; doi:10.1351/pac198855020409.
- ↑ G. Märkl: Phosphabenzol und Arsabenzol. In: Chemie in unserer Zeit 16, 1982, S. 139–148; doi:10.1002/ciuz.19820160503.
- ↑ Christoph Elschenbroich: Organometallchemie. 5. Auflage. Teubner, Wiesbaden 2005, ISBN 3-519-53501-7, S. 220–221.
- ↑ T. Laue, A. Plagens: Namen- und Schlagwort-Reaktionen. 4. Auflage. Teubner, Wiesbaden 2004, ISBN 3-519-33526-3.
- ↑ a b G. P. Elliott, W. R. Roper, J. M. Waters: Metallacyclohexatrienes or Metallabenzenes. Synthesis of Osmabenzene Derivates and X-ray Structure of [Os(C(S)CHCHCHCH)(CO)(PPh3)2]. In: J. Chem. Soc., Chem. Comm. 1982, S. 811–813; doi:10.1039/C39820000811.
- ↑ a b Christoph Elschenbroich: Organometallchemie. 5. Auflage. Teubner, Wiesbaden 2005, ISBN 3-519-53501-7.
- ↑ G. Jia: Progress in the Chemistry of Metallabenzynes. In: Acc. Chem. Res. 37, 2004, S. 479–486; doi:10.1021/ar0201512.
- ↑ Eberhard Breitmaier: Alkaloide: Betäubungsmittel, Halluzinogene und andere Wirkstoffe, Leitstrukturen aus der Natur. 2. Auflage. Teubner Verlag, Wiesbaden 2002, ISBN 3-519-13542-6.
- ↑ G. Löffler, P. E. Petrides: Physiologische Chemie. 4. Auflage. Springer, Berlin 1988, ISBN 3-540-13199-X, S. 69–72.
- ↑ W.Voigt, A. Dietrich, H.-J. Schmoll: Cisplatin und seine Analoga. In: Pharmazie in unserer Zeit 35, 2006, S. 134–143; doi:10.1002/pauz.200500162.
- ↑ M. Galanski, B. K. Keppler: Tumorhemmende Metallverbindungen. In: Pharmazie in unserer Zeit 35, 2006, S. 118–122; doi:10.1002/pauz.200500160.
- ↑ Lehninger: Biochemie. 3. Auflage. Springer, Berlin 2001, ISBN 3-540-41813-X, S. 716–726.
- ↑ G. Löffler, P. E. Petrides: Physiologische Chemie. 4. Auflage. Springer, Berlin 1988, ISBN 3-540-13199-X.
- ↑ W. Kaim, B. Schwederski: Bioanorganische Chemie. 4. Auflage. Teubner, Wiesbaden 2005, ISBN 3-519-33505-0, S. 26–29.
- ↑ a b W. Kaim, B. Schwederski: Bioanorganische Chemie. 4. Auflage. Teubner, Wiesbaden 2005, ISBN 3-519-33505-0.
- ↑ G. N. Schrauzer: Neuere Entwicklungen auf dem Gebiet des Vitamins B12: Reaktionen des Cobaltatoms in Corrin-Derivaten und Vitamin-B12-Modellverbindungen. In: Angew. Chem. 88, 1976, S. 465–474; doi:10.1002/ange.19760881403.
- ↑ H. R. Christen, F. Vögtle: Organische Chemie – Von den Grundlagen zur Forschung. 2. Auflage. Otto Salle Verlag, Frankfurt am Main 1996, ISBN 3-7935-5398-1, S. 14–15.
- ↑ W. Schweisheimer: Parfüms von Tieren. In: Fette, Seifen, Anstrichmittel 67, 1986, S. 381–382.
- ↑ a b H.-D. Belitz, W. Grosch, P. Schieberle: Lebensmittelchemie, 2001, 5. Auflage. Springer-Verlag, ISBN 978-3-540-41096-6, S. 381.
- ↑ a b c d e f D. T. Davies: Basistexte Chemie: Aromatische Heterocyclen. 1. Auflage. Wiley-VCH, Weinheim 1995, ISBN 3-527-29289-6.
- ↑ Jonathan Clayden, Nick Greeves, Stuart Warren: Organische Chemie. 2. Auflage. Springer, Berlin / Heidelberg 2013, ISBN 978-3-642-34715-3, S. 808.
- ↑ a b D’Ans-Lax: Taschenbuch für Chemiker und Physiker. 3. Auflage. Band 1, Springer-Verlag, Berlin-Göttingen-Heidelberg 1967 (ChemieOnline – pKb- und pKs-Werte ( vom 18. Juli 2012 im Internet Archive)).
- ↑ L. Gade: Koordinationschemie. 1. Auflage. VCH, Weinheim 1998, ISBN 3-527-29503-8.
- ↑ K. Banert: Reaktionen ungesättigter Azide, 8. Azidobutatrien und Azidobutenine. In: Chem. Ber. 122, 1989, S. 1175–1178; doi:10.1002/cber.19891220624.
- ↑ C. Paal: Synthese von Thiophen- und Pyrrolderivaten. In: Ber. Dtsch. Chem. Ges. 18, 1885, S. 367–371; doi:10.1002/cber.18850180175.
- ↑ A. Hantzsch: Condensationprodukte aus Aldehydammoniak und ketonartigen Verbindungen. In: Ber. Dtsch. Chem. Ges. 14, 1881, S. 1637–1638; doi:10.1002/cber.18810140214.
- ↑ N. Prileschajew: Oxydation ungesättigter Verbindungen mittels organischer Superoxyde. In: Ber. Dtsch. Chem. Ges. 42, 1909, S. 4811–4815; doi:10.1002/cber.190904204100.
- ↑ Reinhard Brückner: Reaktionsmechanismen. 3. Auflage. Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 138–144.
- ↑ G. Büchi, C. G. Inman, E. S. Lipinsky: Light-catalyzed Organic Reactions. I. The Reaction of Carbonyl Compounds with 2-Methyl-2-butene in the Presence of Ultraviolet Light. In: J. Am. Chem. Soc. 76, 1954, S. 4327–4331; doi:10.1021/ja01646a024.
- ↑ T. Laue, A. Plagens: Namen- und Schlagwort-Reaktionen. 4. Auflage. Teubner, Wiesbaden 2004, ISBN 3-519-33526-3, S. 261–263.
- ↑ Karsten Krohn, Ulrich Wolf: Kurze Einführung in die Chemie der Heterocyclen. Teubner Studienbücher Chemie, 1994, ISBN 978-3-519-03532-9 (Springer-Link).
- ↑ G. Maier, U. Flögel, H. P. Reisenauer, B. Hess, L. J. Schaad: HCl-Abspaltung aus Ethansulfenylchlorid und Chlordimethylsulfid. In: Chem. Ber. 124, 1991, S. 2609–2612; doi:10.1002/cber.19911241134.
- ↑ J. Delepine in: C. R. Hebd. Seances Acad. Sci. 172, 1921, S. 158.
- ↑ Friedrich Asinger und Manfred Thiel: Einfache Synthesen und chemisches Verhalten neuer heterocyclischer Ringsysteme, Angewandte Chemie 70 (1958) 667–683.
- ↑ Jürgen Martens, Heribert Offermanns und Paul Scherberich: Eine einfache Synthese von racemischem Cystein, Angewandte Chemie 93 (1981) 680; Angewandte Chemie-International Edition 20 668 (1981).
- ↑ K Gewald; Schinke, E.; Böttcher, H. Chemische Berichte 99, 1966, S. 94–100.
- ↑ B. Kirste, F. U. Berlin: Substitutionsreaktionen an Aromaten ( vom 4. September 2011 im Internet Archive)
- ↑ D. T. Davies: Basistexte Chemie: Aromatische Heterocyclen. 1. Auflage. Wiley-VCH, Weinheim, 1995, ISBN 3-527-29289-6, S. 37–39.
- ↑ K. P. C. Vollhardt, N. E. Schore: Organische Chemie 3. Auflage. Wiley-VCH, 2000, ISBN 3-527-29819-3, S. 1249–1250.
- ↑ Autorenkollektiv: Organikum. 21. Auflage. Wiley-VCH, 2001, ISBN 3-527-29985-8, S. 395.
- ↑ F. W. Bergstrom, H. G. Sturz, H. W. Tracy: The Use of the Fused Eutectic of Sodium amide and Potassium amide in Organic Synthesis. In: J. Org. Chem. 11, 1946, S. 239–246; doi:10.1021/jo01173a005.
- ↑ J. M. Steward, C. H. Burnside in: Reactions of trimethylenesulfide with chlorine and bromine. In: J. Am. Chem. Soc. 1953, S. 243–244; doi:10.1021/ja01097a517.
- ↑ H. Beyer, W. Walter: Lehrbuch der Organischen Chemie. 23. Auflage. S. Hirzel Verlag, Stuttgart 1998, ISBN 3-7776-0808-4, S. 332.
- ↑ Autorenkollektiv: Organikum. 21. Auflage. Wiley-VCH, 2001, ISBN 3-527-29985-8, S. 302–303.
- ↑ J. Schreiber, D. Felix, A. Eschenmoser, M. Winter, F. Gautschi, K. H. Schulte-Elte, E. Sundt, G. Ohloff, J. Kalovoda, H. Kaufmann, P. Wieland, G. Anner: Die Synthese von Acetylen-carbonyl-Verbindungen durch Fragmentierung von alpha, beta-Epoxy-ketonen mit p-Toluolsulfonylhydrazin. In: Helv. Chim. Acta 50, 1967, S. 2101–2108; doi:10.1002/hlca.19670500747.
- ↑ D. Felix, J. Schreiber, G. Ohloff, A. Eschenmoser: α,β-Epoxyketon: Alkinon-Fragmentierung I: Synthese von exalton und rac – muscon aus cyclododecanon über synthetische methoden. In: Helv. Chim. Acta 54, 1971, S. 2896–2912; doi:10.1002/hlca.19710540855.
- ↑ M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin: An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals. In: Beilstein J. Org. Chem. 7, 2011, S. 442–495; doi:10.3762/bjoc.7.57 (Open Access).
- ↑ a b J. A. Joule, K. Mills: Heterocyclic Chemistry. 4. Auflage. 2000, Blackwell Oxford, ISBN 0-632-05453-0, S. 543–549.
- ↑ Der Sulfolan-Prozess (engl.) ( vom 1. Dezember 2010 im Internet Archive).
- ↑ G. A. Paganini: Heterocycle-based electric conductors. In: Heterocycles 37, 1994, S. 2069–2086.
- ↑ J. Roncali: Conjugated poly(thiophenes): synthesis, functionalisation and application. In: Chem. Rev. 92, 1992, S. 711–738; doi:10.1021/cr00012a009.
- ↑ J. Ferraris, D. O. Cowan, V. V. Walatka, J. H. Perlstein: Electron transfer in a new highly conducting donor-acceptor complex. In: J. Am. Chem. Soc. 95, 1973, S. 948–949; doi:10.1021/ja00784a066.
- ↑ F. Wudl: From organic metals to superconductors: managing conduction electrons in organic solids. In: Acc. Chem. Res. 17, 1984, S. 227–232; doi:10.1021/ar00102a005.